Molecular Formula: C17H19F3N6O
Formula Weight:
380.37
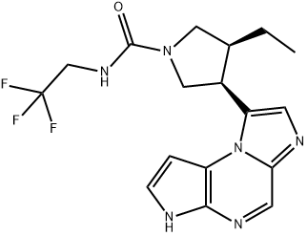
A selective JAK1 inhibitor called upadacitinib was introduced in 2019 by Abbvie for the treatment of active rheumatoid arthritis. It is also being tested in clinical trials for autoimmune conditions like atopic dermatitis, ankylosing spondylitis, and Crohn's disease.
Trifluoroethylurea, a tricyclic imidazolopyrroloxypyrazine core, and (3S,4R) cis-pyrrolidine are among the structural components of upadacitinib. The three steps of the synthetic strategy are as follows: Chiral pyrrolidine was first prepared (Figure 1 molecule L); then it was coupled with pyrroloxypyrazine (Figure 1 molecule C) and then cyclized to produce the imidazole ring; and finally, trifluoroethylurea (Figure 1 molecule Q) was added to produce upadacitinib in quantities of several kilograms.
Steps of the synthetic route are interpreted:
A -> C:Cyclization of arylacetylene A yields the pyrroloxypyrazine heterocycle, which is then in situ protected with toluenesulfonyl chloride to provide the toluenesulfonyl-protected heterocyclic bromide B. Buchwald-Hartwig coupling on acetate is then used to protect the amino group, resulting in the formation of C.

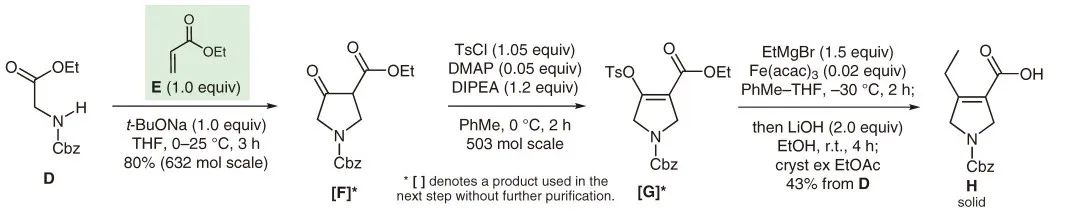
D -> H:Ethyl N-Cbz glycinate D was converted to dihydropyrrolic acid H by condensing it with ethyl acrylate E using t-BuONa to produce the intermediate [F]*. This intermediate was then enolized with TsCl and linked with ethylmagnesium bromide.
H -> K: Ru(OAc)2 [(S)-SEGPHOS] was chosen for scale-up (for commercial availability and ease of use) to produce chiral pyrrolidinoic acid [I]*, which was then smoothly converted to Yelid K by CDI activation of the carboxylic acid and reaction with trimethylsulfoxide. This process involved the evaluation of 48 asymmetric hydrogenation catalysts through high throughput experiments (HTE). No intermediates needed to be separated during the entire process, and the yield was good.

K -> Upadacitinib: Bromomethyl ketone L is produced when H interacts with HBr to form bromomethyl ketone, which is subsequently linked to an amino group-protected heterocycle (C) under basic conditions to form M. M undergoes cyclization upon heating in the presence of trifluoroacetic anhydride (TFAA), resulting in imidazolopyrroloxypyrazine [I]*. The scientists discovered that the organic base had the biggest impact on the pace of cyclization and that pyridine significantly sped up the process. Following deprotection, P was produced, and upadacitinib was produced via a reaction with activated trifluoroethylamine.

Pictures from Synfacts
Refrence:
[1]Development of a Scalable Enantioselective Synthesis of JAK Inhibitor Upadacitinib. Org. Process Res. Dev. 2022, 26, 3, 949–962.
[2]https://www.thieme-connect.com/products/ejournals/pdf/10.1055/s-0041-1738394.pdf